Junior Research Group | Medical Omics
Overview
The Junior Research Group Medical Omics aims to improve healthcare and basic research by providing artificial intelligence-driven tools for the analysis of large biomedical datasets. For comprehensive insights in disease entities, we aim to include multiple layers of data, such as single-cell RNA sequencing*, methylomics data* and medical imaging*. In close collaborations with clinicians at the Charité, we develop support systems for cancer treatment as well as solutions for routine clinical tasks such as automated annotation* of radiological images. Furthermore, we implement feedback systems to utilize expert knowledge in our AI training.
Explanation of terms:
*Single-cell RNA sequencing:
This method measures the expression (simplified: activity) of all genes in a cell in parallel. Modern methods can perform this in parallel for thousands of cells at the same time, for example to study the interaction of immune cells with infected tissue.
*Methylomics data:
Every cell in an organism contains the same genome information. Different cell types in the body thus arise from different activation (expression) of the same genes. The DNA of inactive genes is often chemically altered by methylation. From the measurement of this methylation, the cellular origin of a piece of DNA can be determined.
*Medical imaging:
This term encompasses a variety of procedures, including x-rays, computed tomography (CT), and magnetic resonance imaging (MRI).
*Annotation:
Hand-drawing specific structures in an image.
*Deep neural networks:
This is a machine learning technique in which a mathematical model consisting of multiple layers learns to make predictions based on data. The interconnection of the individual levels of the model bears some resemblance to the communication of individual neurons in the brain.
*Nanopore-based DNA sequencing:
Nanopore-based DNA sequencing determines the sequence of a piece of DNA by pulling it through the pores of a membrane to which a voltage is applied. Since the individual bases ("letters") of the DNA are of different sizes, their electrical resistance changes depending on which base is currently in the pore. From this change in resistance, both the sequence of the DNA and the presence of any chemical modifications can be read.

Topics
Single-cell sequencing
Gene set inference
In contrast to traditional bulk sequencing methods for genomics, single-cell sequencing* gives insights into the workings of a tissue at unprecedented resolution. However, this mass of oftentimes exceedingly noisy and sparse data can be overwhelming when approached with traditional approaches. We have developed a deep-learning-based tool that helps identify functionally related gene sets in single-cell data as well as the cell types and conditions they are related to, allowing researches to more quickly grasp the processes shaping the tissue they are investigating.
COVID-19
During the COVID-19 pandemic, we worked together with other groups at Charité, the University Hospital Leipzig and the DKFZ to deliver some of the first single-cell reference atlases of airways of healthy as well as SARS-CoV-2-positive individuals freely available to the research community, and further used these techniques successfully to demonstrate why some groups of patients are at higher risk of severe COVID-19, while children generally present with less severe disease courses. Beyond scientific curiosity, these projects helped jump-start a phase II clinical trial of a potential drug for the treatment of severe COVID-19.
Other areas
Beyond these two focus areas, we have been involved in a number of different single-cell projects, ranging from spermatogenesis (formation of sperm cells from germ cells) to cardiac dysfunction, as well as in bioinformatics methods development to support novel techniques for the study of individual cells’ transcriptomes.
Glioblastoma
Several ongoing projects are focused on improving diagnosis and management of glioblastoma, the most common and most aggressive primary brain tumor in adults.
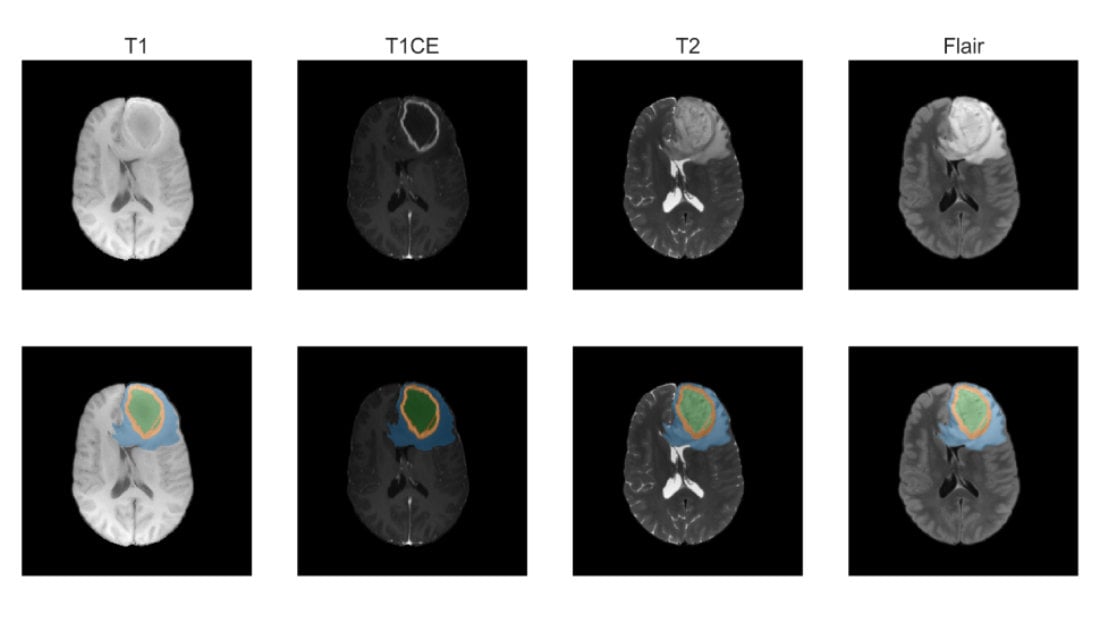
Medical imaging
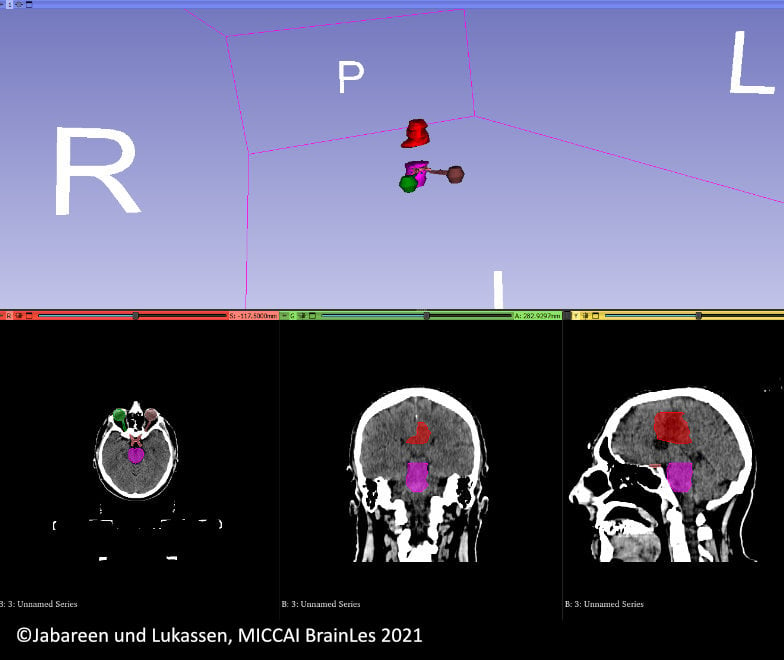
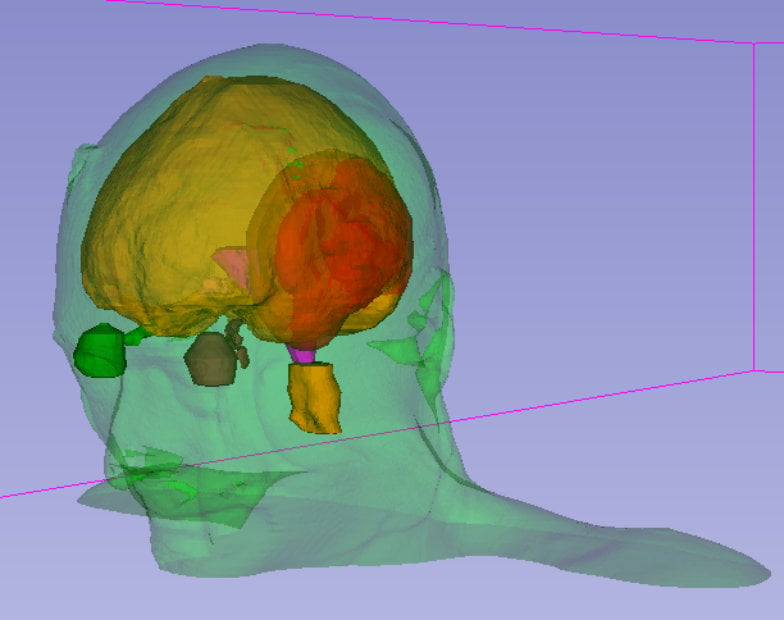
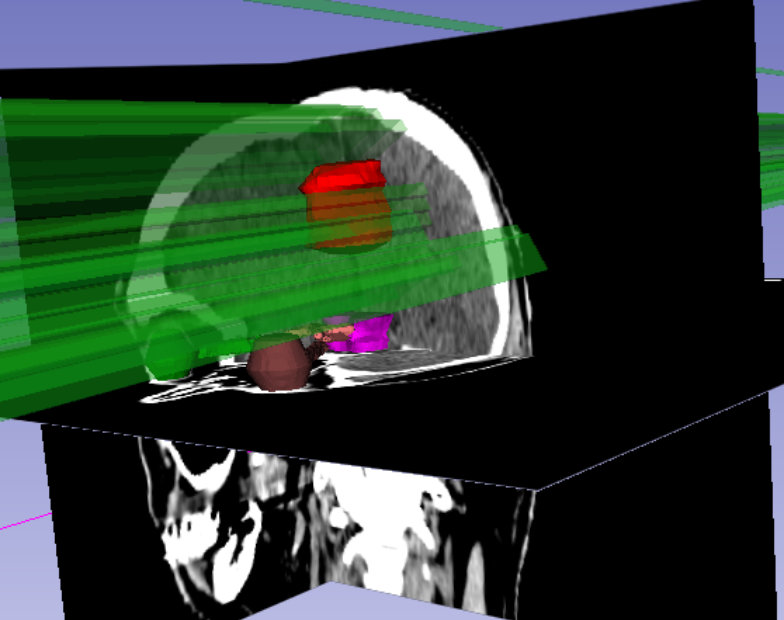
To plan surgery and radiation therapy, the precise location of organs at risk (OARs) and the gross tumor volume (GTV) needs to be determined from 3D MRI or CT scans. Manually annotating these three-dimensional scans is a time-consuming and tedious task. We provide deep neural networks that can accurately segment OARs and GTVs in a matter of seconds, while achieving a performance comparable to human specialist level.
Moreover, we aim to predict the patient survival time with the goal of forecasting the benefit for a patient of receiving a specific radiotherapy regimen while minimizing dose and maximizing efficacy. To ensure the robustness and interpretability of these predictions, we use multiple data sources like radiological features, radiotherapy data, and clinical metadata.
Metholymics
DNA Methylation plays a large role in glioblastoma biology, for example through modulation of the levels of DNA repair enzymes that render a tumor resistant to radiotherapy. Different classes of glioblastomas can be distinguished by their methylation pattern, with relevance for prognosis and treatment.
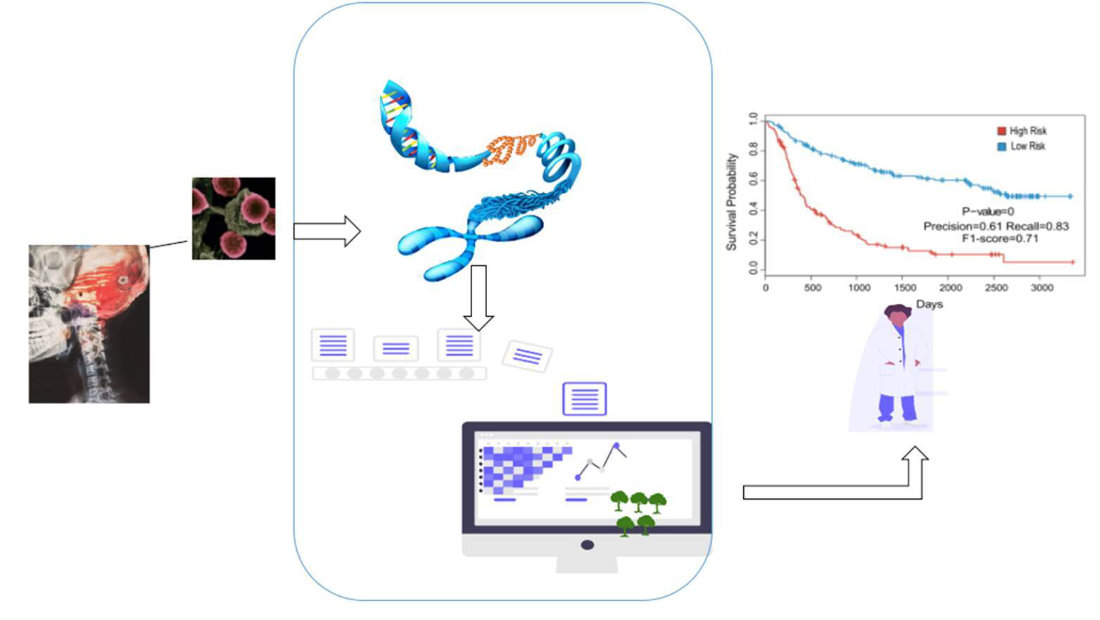
In recent years, shallow nanopore-based DNA sequencing has improved to a level where it can be used to get a reliable, but exceedingly sparse image of DNA methylation within a sample. To bring this technique to the clinics, we work on developing and improving machine learning methods that are equipped to deal with this extreme sparsity and can reliably distinguish different classes of glioblastoma.
Multi-omics
Ultimately, we aim to integrate the results of the different subprojects on glioblastoma, creating a unified framework for diagnosis and management of glioblastoma based on a multitude of data modalities.
Head of the Junior Research Group Medical Omics
Dr. Sören Lukassen
Digital Health Center | Berlin Institute of Health (BIH) at Charité – Universitätsmedizin Berlin
Members of the Junior Research Group Medical Omics
Nabil Jabareen
PhD Candidate | Charité - Universitätsmedizin Berlin
Dongsheng Yuan
PhD Candidate | Charité – Universitätsmedizin Berlin
